FAQ
会社に関するご質問
「真新しいキャンバスに描くようにすべての人が自由に生きられるようにしたい」「私たちはキャンサー・バスターズになりたい」などの当社創業の思いを込めた語「キャンバス」に、「がん治療(Cancer Therapy)に基礎研究(Basic Research)の成果を活かす」という意味を重ねて名付けたものです。
私たちの事業目的は、より良い抗がん剤を一日も早く患者さんにお届けすることです。
この事業目標を実現するための経営理念として、「フェアであること」「科学的・倫理的・経済的に正しい道を最短の距離・時間で進むこと」を常に心掛けます。
また、これらを明確に共有し周知するため、倫理綱領および行動規範を定めています。詳細についてはこちらのページをご覧ください。
この事業目標を実現するための経営理念として、「フェアであること」「科学的・倫理的・経済的に正しい道を最短の距離・時間で進むこと」を常に心掛けます。
また、これらを明確に共有し周知するため、倫理綱領および行動規範を定めています。詳細についてはこちらのページをご覧ください。
私たちは経営方針として「基礎研究と臨床開発の緊密な連携」を重視してきました。
この成果として、CBP501は、一見失敗と見える臨床試験結果の基礎研究チームによる深い解析を契機に、癌細胞のみならず「癌免疫」「癌幹細胞」に関わる広範な作用を有することが明らかになり、免疫系抗がん剤の効果を高める「免疫着火剤」として開発を継続し有望なデータを獲得しつつあります。
こうした活動の結果、少人数組織ながら創薬のすべての段階に対応できる体制を整えることができています。
当社は今後、主にがん免疫領域と免疫系抗がん剤開発で培った知見・経験・ノウハウと、すでに獲得している化合物を活かして、他の領域に眼を向けることなく抗がん剤研究開発を深耕し、最適な開発戦略を選択して「より良い抗がん剤を一日も早く患者さんにお届けする」という経営理念の実現を図ります。
この成果として、CBP501は、一見失敗と見える臨床試験結果の基礎研究チームによる深い解析を契機に、癌細胞のみならず「癌免疫」「癌幹細胞」に関わる広範な作用を有することが明らかになり、免疫系抗がん剤の効果を高める「免疫着火剤」として開発を継続し有望なデータを獲得しつつあります。
こうした活動の結果、少人数組織ながら創薬のすべての段階に対応できる体制を整えることができています。
当社は今後、主にがん免疫領域と免疫系抗がん剤開発で培った知見・経験・ノウハウと、すでに獲得している化合物を活かして、他の領域に眼を向けることなく抗がん剤研究開発を深耕し、最適な開発戦略を選択して「より良い抗がん剤を一日も早く患者さんにお届けする」という経営理念の実現を図ります。
いきさつやメリット・デメリットをブログにまとめています。
こちらをご一読ください。
こちらをご一読ください。
キャンバスの創薬事業・開発・提携に関するご質問
創薬事業には、医薬品開発の過程(詳しくはこちらをご覧ください)において、探索研究の成果を早期に導出(ライセンスアウト)するモデルや、他社で創出され何らかの要因で開発がペンディングとなっている化合物を導入し新たな付加価値を加えて導出するモデルなど、さまざまなビジネスモデルが存在します。
その中で私たちは、自社で探索創出した化合物を臨床開発によって付加価値を十分に高めて製薬企業等に導出する、ハイリスク・ハイリターンのビジネスモデルを志向しています。
特に最先行化合物CBP501に関しては、「創薬パイプライン型」開発を志向する旨を公表しています。
その中で私たちは、自社で探索創出した化合物を臨床開発によって付加価値を十分に高めて製薬企業等に導出する、ハイリスク・ハイリターンのビジネスモデルを志向しています。
特に最先行化合物CBP501に関しては、「創薬パイプライン型」開発を志向する旨を公表しています。
当社の強みは、複数の臨床開発化合物を創出した実績のある独自の創薬プラットフォームと、機動的な基礎研究チーム・臨床開発チーム、それらの緊密な連携を有していることです。
最も先行している化合物CBP501、化合物CBS9106をはじめ、当社の開発する候補化合物はすべて、この創薬プラットフォームから創出され、当社の基礎研究チーム・臨床開発チームの手によって研究開発を推進したものです。
最も先行している化合物CBP501、化合物CBS9106をはじめ、当社の開発する候補化合物はすべて、この創薬プラットフォームから創出され、当社の基礎研究チーム・臨床開発チームの手によって研究開発を推進したものです。
当社の開発品について製薬企業等と提携する際には、製薬企業等との提携による事業収益による開発資金調達や共同開発・開発費負担軽減を原則的な形として取り組んでいます。
一方で、製薬企業等の提携(ライセンス獲得)選好は流動的です。内容にこだわらず提携成立を最優先にすることは、開発リスクやコストの分担と利益配分のバランスの面などで、当社の株主共通の利益を損ないかねません。
また、提携成立後はさまざまな点で、相手先の経営意思決定に大きく左右されるなど、提携・導出のみに依存した企業価値成長戦略には大きな潜在リスクが内在します。
当社は、各パイプラインの状況ごとにそうしたリスクとメリットを勘案し、最適のビジネスモデルによる開発を選択していきます。最先行化合物CBP501については、そのうち「創薬パイプライン型」を指向していることを公表しています。
一方で、製薬企業等の提携(ライセンス獲得)選好は流動的です。内容にこだわらず提携成立を最優先にすることは、開発リスクやコストの分担と利益配分のバランスの面などで、当社の株主共通の利益を損ないかねません。
また、提携成立後はさまざまな点で、相手先の経営意思決定に大きく左右されるなど、提携・導出のみに依存した企業価値成長戦略には大きな潜在リスクが内在します。
当社は、各パイプラインの状況ごとにそうしたリスクとメリットを勘案し、最適のビジネスモデルによる開発を選択していきます。最先行化合物CBP501については、そのうち「創薬パイプライン型」を指向していることを公表しています。
当社と武田薬品工業株式会社は、2010年6月に提携を解消しました。
提携解消時の共同プレスリリースにもあるとおり、現在進めている臨床第2相試験の終了を控え、今後の開発方針に対する考え方について、両社間で大きな相違が見られました。結果として、契約解消が両社にとって最善であるという結論に至り、契約解消を合意するに至ったものです。
当社は、現在の臨床試験で獲得しつつある臨床データを基に、一刻も早く新たな提携パートナーを得て迅速な開発を推進することが、より早期の医薬品上市実現と株主価値向上に寄与すると考え、新たな製薬企業等との提携獲得に向けて積極的な活動をおこなっています。
なお、CBP501臨床試験から得られているデータについて当社と武田薬品工業株式会社は、提携解消時点で「有効性および安全性に関して今後の開発継続の判断を妨げるような事象は出ていない」と見解が一致しています。
提携解消時の共同プレスリリースにもあるとおり、現在進めている臨床第2相試験の終了を控え、今後の開発方針に対する考え方について、両社間で大きな相違が見られました。結果として、契約解消が両社にとって最善であるという結論に至り、契約解消を合意するに至ったものです。
当社は、現在の臨床試験で獲得しつつある臨床データを基に、一刻も早く新たな提携パートナーを得て迅速な開発を推進することが、より早期の医薬品上市実現と株主価値向上に寄与すると考え、新たな製薬企業等との提携獲得に向けて積極的な活動をおこなっています。
なお、CBP501臨床試験から得られているデータについて当社と武田薬品工業株式会社は、提携解消時点で「有効性および安全性に関して今後の開発継続の判断を妨げるような事象は出ていない」と見解が一致しています。
当社とStemline社は2025年6月、両社による話し合いの結果、Stemline社との間でライセンス契約の解消・返還の合意に至りました。
今後当社は、臨床開発段階に進み初期的な有効性・安全性を確認できた抗がん剤候補化合物を追加で実施する基礎研究の成果や会社の財務状況などを勘案して、開発方針を検討していきます。
今後当社は、臨床開発段階に進み初期的な有効性・安全性を確認できた抗がん剤候補化合物を追加で実施する基礎研究の成果や会社の財務状況などを勘案して、開発方針を検討していきます。
当社は、CBP501の提携獲得に向けた活動を日々続けています。
その進捗状況や提携成立時期・規模等の見通しについて、途上の段階で公表はできませんが、その代替として、ぜひ開発の進捗による各パイプラインの魅力向上をご注目ください。
開発の進捗は当社の企業価値向上の根幹であり、それによる魅力の上昇は製薬企業等との提携見通しを予測していただくための最も有力な代替指標です。
その進捗状況や提携成立時期・規模等の見通しについて、途上の段階で公表はできませんが、その代替として、ぜひ開発の進捗による各パイプラインの魅力向上をご注目ください。
開発の進捗は当社の企業価値向上の根幹であり、それによる魅力の上昇は製薬企業等との提携見通しを予測していただくための最も有力な代替指標です。
創薬企業のビジネスモデルのひとつです。
会社プレゼンテーション資料でご説明していますのでご参照ください。
会社プレゼンテーション資料でご説明していますのでご参照ください。
創薬ベンチャー企業の的確な経営判断には、科学的知見の継続的な収集が不可欠であり、当社では、新規化合物の創出と併せ、すでに臨床開発段階にある抗癌剤候補化合物の作用メカニズム研究に注力しています。
厳しい査読のある論文誌への掲載や学会発表を行うことは、当社の研究・開発の品質に係る第三者評価の意味で、重要な意義があると考えています。
厳しい査読のある論文誌への掲載や学会発表を行うことは、当社の研究・開発の品質に係る第三者評価の意味で、重要な意義があると考えています。
業績・財務に関するご質問
当社の単年度業績見通しは、提携契約等の締結に至った場合に当該契約が事業収益・事業費用等に及ぼす影響が極めて大きいと考えられることや、研究開発費に大きな影響を及ぼす臨床試験の進捗について合理的な予測が困難であることから、合理的な業績予想の算定ができないため、公表していません。単年度業績見通しが判明した場合には、速やかにお知らせします。
なお、数字での公表はしていませんが、業績見通しに関する定性的な情報を会社プレゼンテーション資料に掲載しています。
なお、数字での公表はしていませんが、業績見通しに関する定性的な情報を会社プレゼンテーション資料に掲載しています。
当社は創薬事業以外の収益事業を営んでおらず、現在の開発品であるCBP501やCBP9106が将来上市されて得られる収益による黒字化を目指しています。
したがって黒字化の目途は、CBP501やCBS9106の開発・承認・販売のスケジュールに大きく依存します。
この収益の確保に至るまでには一定の期間が必要であることから、研究開発を続けるだけであれば当面の間は赤字基調が続くものと想定されます。
提携契約等の成立に伴う一時金や提携契約に基づくマイルストーン収入等によって一時的に黒字の年度が出現する可能性があり、これら収益の獲得の実現および早期化を目指しています。
したがって黒字化の目途は、CBP501やCBS9106の開発・承認・販売のスケジュールに大きく依存します。
この収益の確保に至るまでには一定の期間が必要であることから、研究開発を続けるだけであれば当面の間は赤字基調が続くものと想定されます。
提携契約等の成立に伴う一時金や提携契約に基づくマイルストーン収入等によって一時的に黒字の年度が出現する可能性があり、これら収益の獲得の実現および早期化を目指しています。
当社は創薬事業以外の収益事業を営んでおらず、現在の開発品であるCBP501・CBS9106等が将来上市されて得られる収益による黒字化を目指しています。
研究開発段階にある当社が計上し得る事業収益は、提携パートナーからの契約一時金・開発協力金・マイルストーン支払金などに限られます。
したがって、製薬企業等の提携パートナーを有していない、または、提携パートナーがあってもアドバイザリー収入などのない期間は、事業収益の計上はありません。
研究開発段階にある当社が計上し得る事業収益は、提携パートナーからの契約一時金・開発協力金・マイルストーン支払金などに限られます。
したがって、製薬企業等の提携パートナーを有していない、または、提携パートナーがあってもアドバイザリー収入などのない期間は、事業収益の計上はありません。
「継続企業の前提に関する注記」(一般に『GC注記』と略称されています)には至らない状況であっても、「売上高の著しい減少」「継続的な営業損失の発生又は営業キャッシュフローのマイナス」など「継続企業の前提に関する重要な疑義を生じさせるような事象又は状況」(一般に『重要事象』と略称されています)にあるときは、有価証券報告書の「事業等のリスク」などにその内容を具体的にわかりやすく開示することになっています。
当社は研究開発段階のベンチャー企業であり、研究開発支出が先行することから、現時点では営業損失や営業キャッシュフローのマイナスが生じることは回避しづらく、この「重要事象」の記載は継続しています。
逆に言えば「継続前提に重要事象」という記載には、当社の場合「営業損失が続いている」以上に深い意味はありません。
当社は研究開発段階のベンチャー企業であり、研究開発支出が先行することから、現時点では営業損失や営業キャッシュフローのマイナスが生じることは回避しづらく、この「重要事象」の記載は継続しています。
逆に言えば「継続前提に重要事象」という記載には、当社の場合「営業損失が続いている」以上に深い意味はありません。
CBP501の将来のスケジュールについては、現時点で予測不能のさまざまな変動要因が存在します。
その上で、理想的かつ最速の開発ができる想定で、「2027年承認・上市」を目指し得ると判断しています。会社プレゼンテーション資料をご参照ください。
その上で、理想的かつ最速の開発ができる想定で、「2027年承認・上市」を目指し得ると判断しています。会社プレゼンテーション資料をご参照ください。
当社は、業績見通しを変更する場合には、適時に当該情報を開示する方針です。
また、各臨床試験について予定被験者数の患者登録が完了した時や臨床試験内容に関する学会発表等の機会には、それぞれ速やかに当該情報を開示し、試験進捗のペース及び内容を適時に正しく周知します。
提携交渉の進捗に関しては、交渉の相手方との守秘義務等も考えられるため、原則として、契約の成立までの期間に細かい経緯を開示する予定はありません。
また、各臨床試験について予定被験者数の患者登録が完了した時や臨床試験内容に関する学会発表等の機会には、それぞれ速やかに当該情報を開示し、試験進捗のペース及び内容を適時に正しく周知します。
提携交渉の進捗に関しては、交渉の相手方との守秘義務等も考えられるため、原則として、契約の成立までの期間に細かい経緯を開示する予定はありません。
決算期は毎年6月末です。
各四半期および通期の決算発表日については、確定次第IRページでお知らせします。なお、予定の変更等やむを得ない事情により、予告なくお知らせと異なる日程で決算の発表を実施することがあります。
最新の決算情報、決算見通しについてはこちら のページをご覧ください。
過去の業績についてはこちら のページをご覧ください。
財務指標についてはこちら こちらのページをご覧ください。
各種レポートや資料についてはこちらこちらのページをご覧ください。
最近のプレスリリース等に関するご質問
CBP501は、これを含む3剤併用投与で、膵臓がん3次治療を対象とする米国FDA監督下の臨床第2相試験で主要評価項目を達成しました。
この内容についてはESMO2023で学会発表済みです。
次相臨床試験については、FDAから臨床第2b相試験の開始承認を得られたほか、現在は欧州での第3相試験開始を図っています。この進捗状況については随時お知らせします。
この内容についてはESMO2023で学会発表済みです。
次相臨床試験については、FDAから臨床第2b相試験の開始承認を得られたほか、現在は欧州での第3相試験開始を図っています。この進捗状況については随時お知らせします。
そのおそれはなく、むしろ、当社が自社で臨床試験費用の全額もしくは大半を負担できることは、早期提携成立の可能性を大きく高めるものと考えています。
現実に、現時点においても、たとえば今後の臨床試験を共同で実施する可能性などを含めた提携パートナー候補企業との交渉を進めており、そのケースにおいては、今般の資金調達が成功した場合に自社で臨床試験費用の全額もしくは大半を負担できる財務基盤を当社が有することは相手方にとってリスク負担の軽減となります。
このように今般の資金調達は、交渉成立の可能性を高めるだけでなく、一日も早い提携パートナー獲得に向けた力強い後押しとなると考えています。
現実に、現時点においても、たとえば今後の臨床試験を共同で実施する可能性などを含めた提携パートナー候補企業との交渉を進めており、そのケースにおいては、今般の資金調達が成功した場合に自社で臨床試験費用の全額もしくは大半を負担できる財務基盤を当社が有することは相手方にとってリスク負担の軽減となります。
このように今般の資金調達は、交渉成立の可能性を高めるだけでなく、一日も早い提携パートナー獲得に向けた力強い後押しとなると考えています。
明確な見通しを申し上げることはできませんが、当社としては、創薬パイプライン型開発を指向しつつ、必要であればできるだけ早いタイミングでの提携パートナー獲得を想定しています。
当社は、インターネット掲示板の投稿をはじめ、当社が発信していない情報については、真偽の確認も含めて、個別にコメントはいたしません。
中には、事実と異なる内容が「キャンバスに問い合わせたら○○と言っていた」などと記載されているケースも少なからず見受けられます。本ウェブサイトやツイッター公式アカウント、各種開示など当社から直接発信した情報(一次ソース)に含まれない情報は、くれぐれも投資判断に利用されないようお願いします。
中には、事実と異なる内容が「キャンバスに問い合わせたら○○と言っていた」などと記載されているケースも少なからず見受けられます。本ウェブサイトやツイッター公式アカウント、各種開示など当社から直接発信した情報(一次ソース)に含まれない情報は、くれぐれも投資判断に利用されないようお願いします。
株式に関するご質問
キャンバスの証券コードは4575です。
100株です。
株価推移についてはこちらをご覧ください。
当社の事業ステージは、現時点では研究開発における先行投資の段階にあるため、株主に対する利益配当及び剰余金配当を実施しておりません。また、今後も当面は、企業体質の強化及び研究開発活動の継続的な実施に備えた資金の確保を優先し、配当は行わない方針です。
株主への利益還元については、当社の重要な経営課題と認識しており、将来的には経営成績及び財政状態を勘案しつつ利益配当及び剰余金配当を検討する所存です。
株主への利益還元については、当社の重要な経営課題と認識しており、将来的には経営成績及び財政状態を勘案しつつ利益配当及び剰余金配当を検討する所存です。
株主総会のスケジュールについてはこちら のページをご覧ください。
株主名簿管理人の三菱UFJ信託銀行株式会社にご連絡ください。連絡先電話番号など詳細についてはこちらをご覧ください。
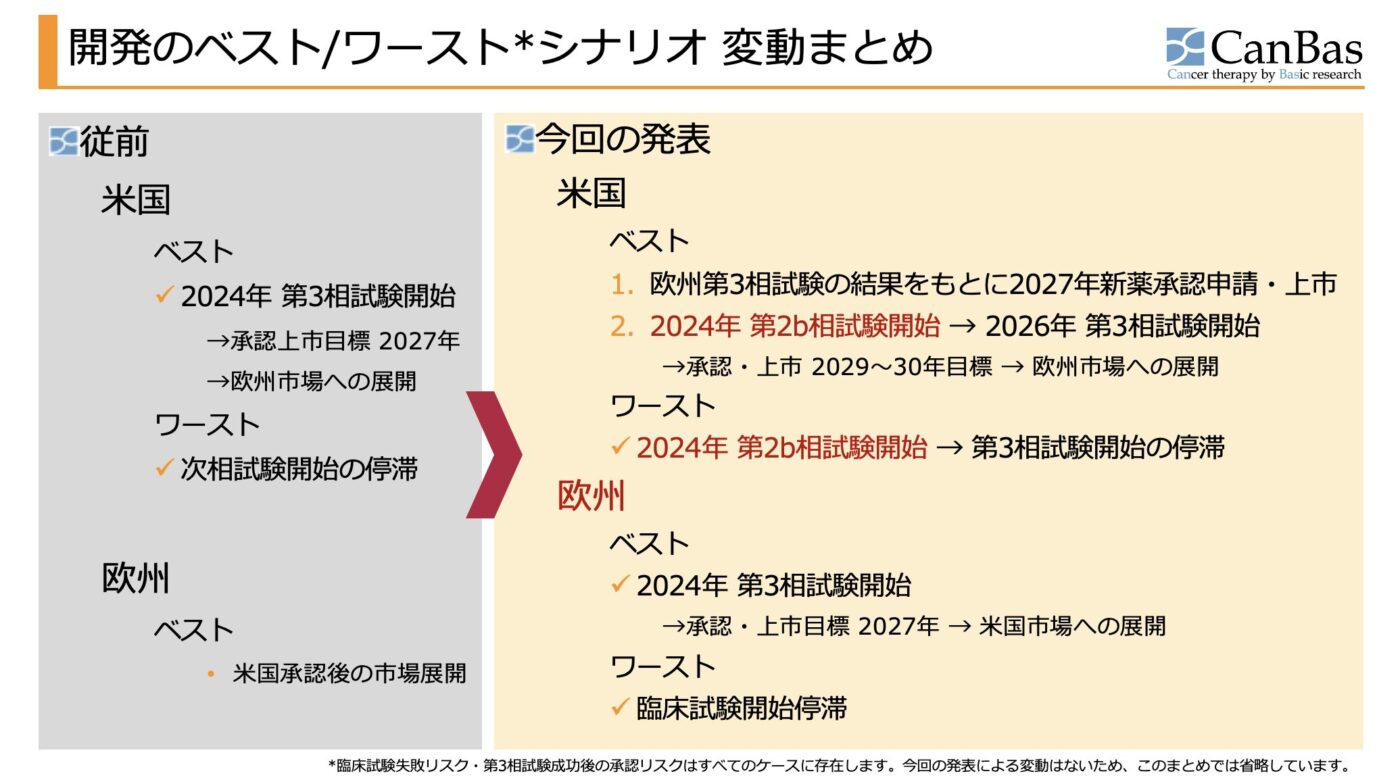
【特設】2024年2月9日公表『CBP501の米国における次相臨床試験予定及び欧州における臨床第3相試験準備について』に関するFAQ(2024年2月11日作成)
今回の公表情報を「CBP501の有効性等が認められず臨床試験が続けられないことが決まった」と同じだという極端に悲観的な見方に対し、それと現状との違いをお伝えしています。
CBP501の有効性等は規制当局に認識され、臨床試験の継続(第2b相試験の開始)が承認されました。 米国で第3相試験に進むという投資家の皆さんの期待の最善や私たちのベストシナリオから外れたのは事実ですが、外れた幅は「臨床試験が続けられない」という状況とは大きく異なります。
また、リスク分散で予め始めていた欧州第3相試験の準備が進んでいて、これが実現すれば所期の目標を達成できる可能性もあります。 この点でも、単に「臨床試験が続けられない」になってしまったのとはまったく異なりますし、ベストシナリオから外れた幅も大きく異なります(外れない可能性もあります)。
これらを出来る限り正確にお伝えした上で、投資家の皆さんそれぞれのご評価を受けたいと考えています。
CBP501の有効性等は規制当局に認識され、臨床試験の継続(第2b相試験の開始)が承認されました。 米国で第3相試験に進むという投資家の皆さんの期待の最善や私たちのベストシナリオから外れたのは事実ですが、外れた幅は「臨床試験が続けられない」という状況とは大きく異なります。
また、リスク分散で予め始めていた欧州第3相試験の準備が進んでいて、これが実現すれば所期の目標を達成できる可能性もあります。 この点でも、単に「臨床試験が続けられない」になってしまったのとはまったく異なりますし、ベストシナリオから外れた幅も大きく異なります(外れない可能性もあります)。
これらを出来る限り正確にお伝えした上で、投資家の皆さんそれぞれのご評価を受けたいと考えています。
米国臨床試験が続けられなくなってしまったわけではなく、第2b相試験への進行自体は承認され、半歩前進しています。欧州にすべて賭けるというわけではありません。
・米国:成功までに要する見込み時間・コストが増加した
・欧州:これまでなかったシナリオが見えつつある
と整理をしていただくと良いと考えています。
・米国:成功までに要する見込み時間・コストが増加した
・欧州:これまでなかったシナリオが見えつつある
と整理をしていただくと良いと考えています。
米国で臨床第3相試験を開始することはできなかった一方で、予め準備を始めておいた欧州試験が実現すれば舞台だけ変わるものの所期の目標を維持できる可能性もある状況でもあり、その両方をご理解いただけるように開示しています。
開始承認はいわば「この内容ならば開始を認めますが、どうしますか?」という、協議の流れの中での通知のひとつです。受領の時点では、当社がそのまま受け入れるかどうか決めておらず、この開始承認を受け入れず協議を継続することも選べる状態です。
今回の開示は、あらかじめリスクヘッジとして準備を始めておいた欧州臨床試験申請の準備状況などを含めて検討し、これ以上協議を継続せず開始承認内容を受け入れる旨を取締役会で決定したので、決定事実としてすみやかに公表したものです。
今回の開示は、あらかじめリスクヘッジとして準備を始めておいた欧州臨床試験申請の準備状況などを含めて検討し、これ以上協議を継続せず開始承認内容を受け入れる旨を取締役会で決定したので、決定事実としてすみやかに公表したものです。
こちらの図表をご参照ください。
私たちは、
(1) 第3相試験ではないものの、臨床試験の継続(第2b相試験の開始)は承認されたことで、米国で開発を継続する場合のワーストシナリオが底上げされた
(2) 予め準備を始めておいた欧州試験によって、ベストシナリオの可能性が維持された
と考えています。

私たちは、
(1) 第3相試験ではないものの、臨床試験の継続(第2b相試験の開始)は承認されたことで、米国で開発を継続する場合のワーストシナリオが底上げされた
(2) 予め準備を始めておいた欧州試験によって、ベストシナリオの可能性が維持された
と考えています。
臨床第2相試験の結果得られた3剤併用投与の有効性等を示唆するデータについて、規制当局が懐疑的ということはありません。実際に、第3相試験ではないものの第2b相試験の開始が承認されています。
また、第2相試験のプロトコールはそれ以前にFDAと協議して策定したものであり、これに対する不備等の指摘もありませんでした。第2相試験でステージ2を実施しなかったことについても同様です。
また、第2相試験のプロトコールはそれ以前にFDAと協議して策定したものであり、これに対する不備等の指摘もありませんでした。第2相試験でステージ2を実施しなかったことについても同様です。
はい、米国での第3相試験開始に向けた協議は一旦終了です。第2b相試験(もし実施するとすればですが)を実施しつつ第3相試験へ向けた協議を再開することになります。
特別な状況においても一切ないというわけではありませんが、現時点でそれを強調するのは適切でなく、ないとお考えいただくのが妥当と考えています。
今回協議され結論に至らなかった内容をやや単純化してご説明します。
A・B・Cの3剤があります。どれも単独では、既存治療(医師の最適選択)と比べてほぼ効きません。 一方、A+B+Cの3剤併用は、既存治療と比べて統計的に証明できるほどの効果が期待できます。
この3剤併用を新しい治療法として承認するためには、原則として新薬Cの寄与(A+Bの2剤併用との差)を証明することになっています。 そのために規制当局は、「A+B+C」と「A+B」の2群比較試験を指示するべきです。
しかし、A+Bが効くかもしれないとする過去の臨床試験はありません。さらに、ひとつ前の臨床試験でその群は臨床医師たちによって「当該投与群に被験者を登録することはできない」と推奨されました。A+Bの投与群を設定する臨床試験は、被験者の利益に鑑みて倫理的に問題があり、規制当局としては指示できません。
この「指示するべき」「指示できない」の両面ある2剤併用投与群をどうするのか、長い協議をしてきたのですが、今回は規制当局が結論を出しませんでした。
ただ、冒頭に戻ると、A+B+Cは既存治療と比べれば統計的に差を証明できるくらい有効な可能性があります。現在治療法のない患者の皆さんの利益からすると、A+B+Cは新治療法として高い価値があるかもしれません。それをどうやって証明するのかは、ESMO2023の膵臓がんセッションでも「今後さらに最適解を見つける努力を(医学界や製薬業界が)続ける価値がある」と指摘されていました。
今回、残念ながらFDAは結論表明に至りませんでしたが、どちらに結論が転んでも不思議のない協議でした。 ちなみに私たち自身は、結論が決まりさえすればどちらでも対応することができます。
A・B・Cの3剤があります。どれも単独では、既存治療(医師の最適選択)と比べてほぼ効きません。 一方、A+B+Cの3剤併用は、既存治療と比べて統計的に証明できるほどの効果が期待できます。
この3剤併用を新しい治療法として承認するためには、原則として新薬Cの寄与(A+Bの2剤併用との差)を証明することになっています。 そのために規制当局は、「A+B+C」と「A+B」の2群比較試験を指示するべきです。
しかし、A+Bが効くかもしれないとする過去の臨床試験はありません。さらに、ひとつ前の臨床試験でその群は臨床医師たちによって「当該投与群に被験者を登録することはできない」と推奨されました。A+Bの投与群を設定する臨床試験は、被験者の利益に鑑みて倫理的に問題があり、規制当局としては指示できません。
この「指示するべき」「指示できない」の両面ある2剤併用投与群をどうするのか、長い協議をしてきたのですが、今回は規制当局が結論を出しませんでした。
ただ、冒頭に戻ると、A+B+Cは既存治療と比べれば統計的に差を証明できるくらい有効な可能性があります。現在治療法のない患者の皆さんの利益からすると、A+B+Cは新治療法として高い価値があるかもしれません。それをどうやって証明するのかは、ESMO2023の膵臓がんセッションでも「今後さらに最適解を見つける努力を(医学界や製薬業界が)続ける価値がある」と指摘されていました。
今回、残念ながらFDAは結論表明に至りませんでしたが、どちらに結論が転んでも不思議のない協議でした。 ちなみに私たち自身は、結論が決まりさえすればどちらでも対応することができます。
医薬品の規制に関する国際的調和が進められており、他の地域の規制当局の監督下で実施された臨床試験(この場合は第2相試験まで)で得られたデータや知見を申請に活用することが可能な制度になっています。
欧州第3相試験の準備状況は、規制当局への申請に必要な事前作業(EMAは事前提出書類等が米国よりもたくさんあります)がほぼ終わり、正式な臨床第3相試験開始申請を提出予定という段階です。
2024年2月27日更新
欧州における規制当局EMA(欧州医薬品庁)への臨床試験開始申請⼿続きを開始し、当社が実施を希望している臨床第3相試験の具体的な内容に関する協議が始まりました。こちらのリリースをご参照ください。
2024年2月27日更新
欧州における規制当局EMA(欧州医薬品庁)への臨床試験開始申請⼿続きを開始し、当社が実施を希望している臨床第3相試験の具体的な内容に関する協議が始まりました。こちらのリリースをご参照ください。
臨床試験のプロトコール等に関する個別具体的なミーティング等は臨床試験開始申請提出からスタートします。現在は、その申請に必要な事前作業がほぼ終わった状態です。したがって現時点では正式な手続きとしての事前協議等を当局と実施しているわけではなく、欧州臨床試験で承認獲得実績のある研究者・医師らとの協働の中で手応えを持つに至っています。
2024年2月27日更新
欧州における規制当局EMA(欧州医薬品庁)への臨床試験開始申請⼿続きを開始し、当社が実施を希望している臨床第3相試験の具体的な内容に関する協議が始まりました。こちらのリリースをご参照ください。
2024年2月27日更新
欧州における規制当局EMA(欧州医薬品庁)への臨床試験開始申請⼿続きを開始し、当社が実施を希望している臨床第3相試験の具体的な内容に関する協議が始まりました。こちらのリリースをご参照ください。
今回、米国規制当局との間で結論に到達しなかったのは、科学的・統計的な議論ではなく、多剤併用投与をどのような証明で承認するかに関する規制当局の方針の話です。
したがって、当局によって考え方や基準が異なっても不思議はありません。
実際にさまざまな点で米国と欧州は異なる判断をしており(具体的な事例については他社の具体的な会社名等になってしまうので、慎重に実施していきます)、多少強引でもそこをこじ開けに行く価値はあると考えています。
したがって、当局によって考え方や基準が異なっても不思議はありません。
実際にさまざまな点で米国と欧州は異なる判断をしており(具体的な事例については他社の具体的な会社名等になってしまうので、慎重に実施していきます)、多少強引でもそこをこじ開けに行く価値はあると考えています。
臨床第2相試験の内容は、単にFDAと合意したというだけのものではなく、科学的統計的に適切であることをFDAも事前に確認した内容です。
したがって、そこから得られたデータの解釈等についてEMAもさほど変わらない判断になると考えていますが、ご指摘のとおりそのおそれはあり、その場合は協議の経過や結論の内容によって検討します。
したがって、そこから得られたデータの解釈等についてEMAもさほど変わらない判断になると考えていますが、ご指摘のとおりそのおそれはあり、その場合は協議の経過や結論の内容によって検討します。
「2027年承認・上市」の目標を変えていないのが私たちの「意気込み」の表明です。試験の規模にもよりますが、2024年末前後までに臨床試験を開始し、目標の堅持を投資家の皆様にも目に見える形でお示ししたいと考えています。
臨床試験期間の長短は、登録患者数や実施施設数などによって大きく変動します(「2024年6月」の説明にもその旨を付記しています)。開始申請を予定している試験内容がEMAに承認されれば、2024年末前後までの組入れ開始で目標を維持できると考えています。
はい、そう考えています。何より大きいのはESMO2023で臨床第2相試験結果の学会発表が済んでいることで、協議においてはそれが力強い支援材料になると期待しています。
はい、もちろんこれまでの臨床試験結果は変わりません。また、これまでの臨床データを元に(第3相ではないものの)次の臨床試験の開始が承認されていますから、FDAとの交渉前そのままではなく、「規制当局が有効性の兆候を認め臨床試験の継続を承認した」という、半歩進んだ状況にあります。
欧州で第3相試験を開始できた場合の良い点としては、同じ欧州で開催されたESMO2023でご注目いただいたことがダイレクトに現場の医師へ伝わっているので(欧州と米国は一体のようで、そういう点で微妙な差がありがちです)、医師の協力的・積極的な姿勢によって円滑な患者組入れが米国以上に期待できることなどが挙げられます。
はい、新たに契約が必要です。ただ、CROに対する支払いの多くは被験者登録以降の費用なので、もし米国第2b相試験直前まで準備を進めておいてペンディングする場合は今後約2.5億円の支払いになります。
公表資料でも触れているとおり、米国での開発継続となった場合には迅速に再開できるよう、念のため米国臨床第2b相臨床試験の準備も進めておきます。
実施しなくなった場合にはおっしゃるようにロスになり、決して小さな金額ではないのですが、「保険」として意味のある支出と判断しています。
実施しなくなった場合にはおっしゃるようにロスになり、決して小さな金額ではないのですが、「保険」として意味のある支出と判断しています。
欧州第3相試験が成功したときは、そのデータに基づく米国新薬承認申請実施を想定しています。
過去の事例を見ても高低さまざまで一概には言えませんが、ざっくり言ってしまうと「第3相試験で示されたデータの度合いによる」というのが要因のひとつと考えています。規制当局は「証明されていないものを認めない」と同時に「証明された新薬は逃さず認めたい」ものでもあります。
時間がかかったことについては私たち自身大変残念に思っていると同時に、投資家の皆さんのご不満はそれ以上であられると思います。リスク分散として準備してきた欧州臨床試験の早期実現を目指します。
「創薬パイプライン型」は何が何でも自社で最後まで進めることに意固地にこだわるものではなく、以前から資料でお示ししているとおり、状況に応じて選択していくビジネスモデルです。
このモデルの長所は、順調に成功に至ったときに株主の利益を最大化できることだけでなく、順調でないときも含めて、開発の意思決定を他社に影響されず自社のみでコントロールできることです。
今回、もしCBP501を早々に導出した状態で今回と同様の規制当局協議結果となっていたら、当社の中長期的な企業価値を最優先した意思決定となったかどうかまったくわかりません。欧州臨床試験への展開による上市目標の維持などを検討されることなく、提携解消など厳しい事態になっていたかもしれません。少なくとも今回は、自社の中長期的な企業価値最大化を最優先した意思決定ができたことや、それについてこうやって投資家の皆さんと質疑応答ができ今後の展開を議論できることは、CBP501を創薬パイプライン型で進めてきた選択が良いほうに出たと考えています。
今後も常時柔軟に、中長期的な企業価値を最大化するためにより有利なほうを選択していきます。
なお現状については、第3相試験への準備が欧州で続いているほか、それが実現しない場合は米国第2b相試験にもシフトできる状況です。重要な選択が多数続くので、意思決定の主導権を確保できる創薬パイプライン型開発のメリットが比較的大きい時期に差し掛かっていると考えています。
このモデルの長所は、順調に成功に至ったときに株主の利益を最大化できることだけでなく、順調でないときも含めて、開発の意思決定を他社に影響されず自社のみでコントロールできることです。
今回、もしCBP501を早々に導出した状態で今回と同様の規制当局協議結果となっていたら、当社の中長期的な企業価値を最優先した意思決定となったかどうかまったくわかりません。欧州臨床試験への展開による上市目標の維持などを検討されることなく、提携解消など厳しい事態になっていたかもしれません。少なくとも今回は、自社の中長期的な企業価値最大化を最優先した意思決定ができたことや、それについてこうやって投資家の皆さんと質疑応答ができ今後の展開を議論できることは、CBP501を創薬パイプライン型で進めてきた選択が良いほうに出たと考えています。
今後も常時柔軟に、中長期的な企業価値を最大化するためにより有利なほうを選択していきます。
なお現状については、第3相試験への準備が欧州で続いているほか、それが実現しない場合は米国第2b相試験にもシフトできる状況です。重要な選択が多数続くので、意思決定の主導権を確保できる創薬パイプライン型開発のメリットが比較的大きい時期に差し掛かっていると考えています。
ご質問のように欧州で第3相試験が始まった場合には、そのデータを使った米国申請も可能なので、わざわざ米国の臨床第2b相を引き継ぐ提携は考えづらいです。その場合はワールドワイドの提携を検討することになります。
欧州第3相試験が実現した場合にはその結果に基づく米国新薬承認取得も想定しており、現時点で当社の主張するパイプライン価値試算の前提数値(米国のみの想定)の変更は考えていません。